
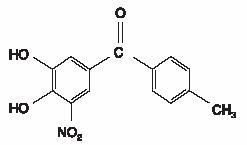
Tolcapone (Tasmar)
Trade Name : Tasmar
Valeant Pharmaceuticals North America LLC

TABLET, FILM COATED
TOLCAPONE Catechol O-Methyltransferase Inhibitors [MoA],Catechol-O-Methyltransferase Inhibitor [EPC]
Delivery Process

Submit a Request


We’ll Get in Touch

Confirmation and Payment
Submit a Request
Product information is meant for
Disclaimer
Trade Marks displayed in compliance with provisions of: Trademark Act, 1999 u/s 30 and 30 (1) of "Fair use"
GNH India is WHO GDP and ISO 9001 2015 Certified Pharmaceutical Wholesaler/ Supplier/ Exporters/ Importer from India of Tolcapone (Tasmar) which is also known as Tasmar and Manufactured by Valeant Pharmaceuticals North America LLC. It is available in strength of 100 mg/1 per ml. Read more 
Tolcapone (Tasmar) is supplied for Tenders/ Emergency imports/ Un - licensed, Specials, Orphan drug/ Name patient line/ RLD supplies/ Reference listed drugs/ Comparator Drug/ Bio-Similar/ Innovator samples For Clinical trials. Click to know price. Read less
Packaging and Delivery

Validated Cold Chain Shipment
We deliver your medicines through a validated cold chain shipment process. This process is used as these medicines need to manufactured, transported and stored at very specific temperatures, utilizing thermal and refrigerated packaging methods.
Inquire directly from our website and get 5% off on any medicine!
We deliver your medicines through a validated cold chain shipment process. This process is used as these medicines need to manufactured, transported and stored at very specific temperatures, utilizing thermal and refrigerated packaging methods.
Read moreAbout GNH
We deliver your medicines through a validated cold chain shipment process. This process is used as these medicines need to manufactured, transported and stored at very specific temperatures, utilizing thermal and refrigerated packaging methods.
We deliver your medicines through a validated cold chain shipment process. This process is used as these medicines need to manufactured, transported and stored at very specific temperatures, utilizing thermal and refrigerated packaging methods.
Read more










 Disclaimer
Disclaimer

